Advances in single-cell RNA sequencing (scRNA-seq) have dramatically enhanced our understanding of cellular functions and disease mechanisms. Despite its potential, scRNA-seq faces significant challenges related to data privacy, cost, and Intellectual Property (IP) protection, which hinder the sharing and collaborative use of these sensitive datasets. In this paper, we introduce a novel method, scE(match), a privacy-preserving tool that facilitates the matching of single-cell clusters between different datasets by relying on scmap as an established projection tool, but without compromising data privacy or IP. scE(match) utilizes homomorphic encryption to ensure that data and unique cell clusters remain confidential while enabling the identification of overlapping cell types for further collaboration and downstream analysis. Our evaluation shows that scE(match) performantly matches cell types across datasets with high precision, addressing both practical and ethical concerns in sharing scRNA-seq data. This approach not only supports secure data collaboration but also fosters advances in biomedical research by reliably protecting sensitive information and IP rights.
Introduction
Single-cell transcriptomic data provides a high-dimensional view into gene-expression patterns at single-cell resolution from human tissue and model systems [1][2][3]. Recent advances in this area [4], most notably, fast and scalable single-cell RNA sequencing (scRNA-seq) technology, contribute to a better understanding of the human body and its disease mechanisms. Such high-dimensional and large-scale data availability has also revolutionized computational biology [5], where novel tools are being developed for data management, processing, and downstream analyses. Here, unsupervised clustering cells with a similar expression fingerprint enables the identification of cell clusters with specific cell molecular functions and properties. These cell types can be further classified into phenotypically different cell states, which can be linked to diseases such as COVID [6], cancer [7], myocardial infarction [8], heart [1] or chronic kidney disease [2][3][9].
As scRNA-seq data contains patient-specific gene-expression information, it is sensitive data; in fact, scRNA-seq data can be subject to similar threats as classic genetic data [10] with data volumes being orders of magnitude larger due to cell-level resolution. At the same time, scRNA-seq technology is still expensive to operate [11], with individual experiments requiring consumables worth tens of thousands of Euros besides expensive sequencing equipment. Researchers thus seek to share and reuse datasets as much as possible [12]; however, the above data privacy concerns significantly limit the willingness and opportunities for sharing data.
Data privacy is not the only concern when sharing or collaborating on scRNA-seq data. Specialized labs invest significant resources into collecting and analyzing scRNA-seq data [11], and the proper attribution of findings from analyzing such data represent thus is crucial to ensure sustainable funding for future experiments. The Intellectual Property (IP) of collaborating laboratories thus must be preserved, including newly identified cell functions that have not yet been published. However, reducing redundancies, consolidating expertise for early comparison of results, and bootstrapping collaboration are necessities to minimize costly parallel research efforts. Besides, such a comparison would also help to agree on common cell type and cell state annotation schemes early on, thereby increasing the findability and accessibility of results (cf. FAIR principles [13]). Possibly due to the IP concerns, such collaboration is a rare current practice. Hence, knowledge gained from the data complements the sensitive features, and both require effective data protection.
One common problem in scRNA-seq data analysis that allows for alleviating this situation is the projection of query datasets into other reference datasets [14], for instance, to validate that larger reference datasets exhibit similar cell characteristics (identified by clustering). Unfortunately, currently established tools [15][16][17][18] all require direct access to both datasets, rendering them unsuitable for inter-organizational collaboration. In this work, we mitigate this problem by building upon an established, well-known projection tool called scmap [15] and propose a privacy-preserving adaptation of it. Specifically, we introduce scE(match), which securely matches clustered cells under homomorphic encryption. Compared to the query to reference dataset mapping by scmap, scE(match) matches datasets of two entities in either direction, thereby identifying correspondences across remote datasets. These correspondences enable researchers to identify common discoveries, e.g., to initiate further collaboration, data standardization, and find orthogonal evidence for their discoveries in independent datasets. At the same time, scE(match) does neither reveal the dataset nor identified cell clusters exclusive to a single organization.
Contributions Our primary contributions are as follows.
Based on state of the art, scmap [15], we develop scE(match)—our privacy-preserving adaptation—for matching single-cell clusters between two parties to identify overlapping cell types while meeting the confidentiality and intellectual property requirements of data subjects and labs.
Our performance evaluation of scE(match) demonstrates its feasibility in large real-world research settings, promising immediate benefits for data subjects and labs alike.
Based on two state-of-the-art single-cell datasets, we show that scE(match)’s calculated matchings are of practical relevance and are indeed suitable to initiate further collaboration.
We open source our implementation at https://github.com/COMSYS/scEmatch to foster further research and development in the field of privacy-preserving single-cell data analysis.
Organization The remainder of this paper is structured as follows. Section 2 provides the necessary background on single-cell data and introduces the current state of the art in single-cell data analysis besides current privacy-related challenges in this area. Section 3 then details the research gap and outlines the problem statement. Section 4 introduces the necessary building blocks for our privacy-preserving cluster-matching approach. Section 5 details the design of scE(match), our privacy-preserving adaptation of scmap. Section 6 evaluates the performance and precision of scE(match) using real-world single-cell data. Section 7 discusses the security implications of our approach. Finally, Section 8 concludes the paper and outlines future work.
Background and Related Work
This section outlines essential background knowledge on single-cell data, reviews the current state of the art in single-cell data analysis, and discusses privacy challenges in this field.
Single-Cell Data and Cluster Matchings
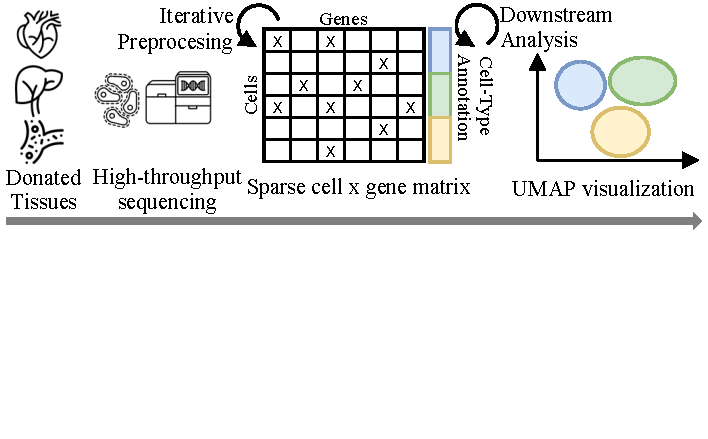
Significant advances in high-throughput sequencing technologies in the past decade have enabled the mass collection of single-cell data, i.e., biological information such as RNA, DNA, protein, or chromatin data at the single-cell level [19] Single-cell data thus is in the process of revolutionizing the field of biology and medicine [20] and has already led to a better understanding of biological mechanisms at cell-level [1][2][3]. Figure 1 visualizes a typical analysis pipeline. Depending on the tissue type, data is collected from living or deceased individuals who donate tissues for research. For instance, blood samples are taken from living donors, whereas heart tissues are typically only available post-mortem, resulting in limited availability. Single-cell data analysis then builds upon a complex and computationally demanding pipeline, including quality control, normalization, imputation, data integration, and dimensionality reduction as preprocessing steps, followed by downstream analysis tasks for the discovery of biological insights [21][20]. These insights comprise identifying cell types or functions, cell-to-cell communication, disease trajectories, and beyond [20]. Here, Molho et al. [20]provide an extensive overview of the computational and data-scientific aspects of single-cell data analysis, and we refer to their work for further details [20]. In this work, we focus on single-cell RNA sequencing (scRNA-seq) data, which is among the most common single-cell data types [20].
scRNA-seq data is typically represented as a sparse cell$\times$gene matrix (cf. Figure 1), with rows corresponding to cells and columns to genes. The challenge then is to separate meaningful differences in gene expression between cells from technically induced noise [22], batch effects, and other artifacts, such that downstream analysis like cell-type annotation and clustering can focus on biologically meaningful gene expressions only. Due to the high dimensionality of this data (thousands of genes per cell and thousands of cells per donor), such insights are typically visualized in a reduced two-dimensional space such as UMAP embeddings [21].
To compare identified functional clusters between different datasets, each prone to batch effects and further measurement artifacts, data integration is a crucial task in single-cell analysis. Here, one can project the clusters of one dataset into the space of the other dataset, e.g., using scmap [15], FR-Match [16], or other data integration methods like Harmony [17] or Seurat [23]. All these methods, however, require direct access to both datasets [24].
Related Work
In the following, we introduce relevant works for integrating single-cell data besides privacy challenges and privacy-preserving processing options centering around single-cell data.
Single-Cell Data Integration Tools Several tools exist for integrating single-cell datasets from heterogeneous sources that employ different approaches to integrate cells, clusters, or whole datasets [15][16][17][25][24][26][18]. scmap [15] relies on a nearest neighbor search for either cells or cluster centroids and employs cosine similarity, Pearson correlation, and Spearman correlation to assess similarity. FR-Match [16], in contrast, employs supervised feature selection to detect marker genes for specific cell types and performs graph-based matching to integrate datasets. Then again, Harmony [17] iteratively computes shared linear embeddings while optimizing for clusters of cells from different sources. These linear projection methods are complemented by deep-learning approaches like scVI [25], scANVI [24], scGen [26], and recently, scGPT [18], which employ deep neural networks to integrate datasets, among other tasks. When using integrated datasets for downstream analysis, Harmony performs well for simpler datasets, while deep learning approaches are more suitable for more complex tasks [18]. Nonetheless, computationally-cheaper tools like scmap still perform well for comparing clustered datasets [16].
Issues in Privacy-Preserving Processing Single-Cell Data While the steady increase in the availability of single-cell data has led to a surge in research, privacy issues centering around single-cell data have been mainly ignored. The contained genetic fingerprint contained in these samples is highly sensitive, encompassing information on ancestry and ethnicity [27][28], enabling the deanonymization of individuals in large association studies [10], or from public databases [29]. To the best of our knowledge, none of the existing privacy issues have been addressed explicitly in the context of single-cell data; however, as scRNA-seq data contains the same genetic information at a much higher volume, we suppose that one has to expect similar privacy issues here. In this vein, multiple works have been proposed to address privacy concerns in single-cell data analysis, including scFed [30], scPrivacy [31], and PPML-Omics [32]. These works employ federated learning principles for clustering scRNA-seq data but lack comparison functionality. Others have employed homomorphic encryption for genomic disease testing [33] or principles from multiparty computation for genome-wide association studies [34].
Privacy-Preserving Data Science Beyond single-cell analysis, privacy-preserving clustering algorithms have been proposed for other applications in various domains, e.g., k-means based on two-party computation with oblivious transfer [35], multi-party dbSCAN [36], or differentially-private clustering via hierarchically separated trees [37]. These works show the general suitability of these building blocks for clustering tasks, albeit with significant performance penalties [35]. However, to the best of our knowledge and despite the urge for such a tool, no privacy-preserving clustering comparison tool has yet been proposed for single-cell data analysis.
Problem Statement
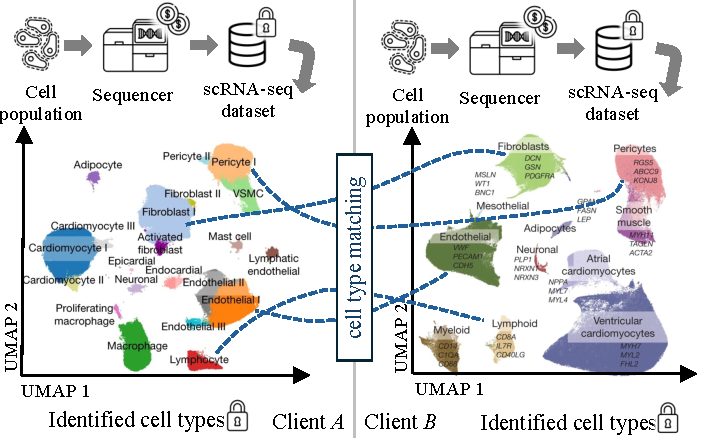
Consider two research labs, independently collecting scRNA-seq data from different donors, and each identifying cell clusters that are of interest to them, as shown in Figure 2. Both labs, to which we refer as Clients $A$ and $B$, are interested in comparing their findings to identify overlapping cell types, which could indicate common cell functions or disease mechanisms. However, both labs are also interested in keeping their datasets and discoveries confidential to maintain their individual advances. Besides, the GDPR-related privacy requirements of donors often obligate labs not to share this sensitive genetic information at all [38]. As such, clients are reluctant or incapable of sharing their datasets directly with each other, as this would reveal their data and identified cell clusters to the other party, besides violating the data privacy of donors. In the following, we formalize our threat model addressing this situation and discuss that related work fails to address this issue before deriving design goals that our solution to this problem, scE(match), must meet.
Threat Model As genetic data is genetically linkable to individuals [10] while containing sensitive information like disease markers [27], the patient-level data must be kept confidential, i.e., remain inaccessible to third parties. Due to the data providers’ regulatory compliance, it is also in their interest that third parties may not infer any privacy-relevant information from the data they process. Thus, we consider everyone but the initial data-collecting entity (i.e., the client) as a third party, including the other client holding the data one aims to compare against and any computational infrastructure not under the control of the data-collecting entity. These third parties can inspect any received data and metadata and might have background knowledge about the data to learn as much as possible from processed data. At the same time, they will not modify any data or interfere with the computations as remote clients are also interested in results and thus provide meaningful input data. Besides, we suppose that infrastructure operators have no incentive to violate data integrity: They are interested in maintaining their reputation and providing accurate results if they receive monetary compensation for calculation. We thus assume all entities to be honest-but-curious [39].
Research Gap Single-cell mapping and integration tools like scmap [15] or Harmony [17] have shown that comparing scRNA-seq data is feasible and beneficial for research. However, these tools are currently unsuitable for inter-organizational collaboration, as they require direct access to both datasets, which does not meet the above-outlined confidentiality requirements and intellectual property concerns of the data providers. Thus, the research question of how to facilitate such collaboration while preserving the privacy of data providers remains unresolved. Among the existing mapping tools, scmap [15] and Harmony [17] rely on computationally simpler methods compared to deep learning approaches like scVI [25], which makes them better-suited for adaptation to confidentiality-preserving computing methods, such as those methods used previously for privacy-preserving clustering in other application domains (cf. Section 2.2).
Design Goals Based on the outlined research gap and threat model, we derive the following design goals for scE(match):
Data Privacy: Ensure that sensitive genomic data of donors remains confidential and inaccessible to third parties.
Matching Confidentiality: Ensure that the identified cell clusters of the data providers are not accessible to anyone else, such that the intellectual property of the data providers is protected.
Matching Utility: Ensure that the matching results are of similar quality as those of scmap, such that the data providers can source the comparison of their datasets to bootstrap collaboration.
Scalability: Ensure that the matching process scales to large datasets, as prevalent in related work, such that the data providers can compare their datasets efficiently.
Preliminaries
In this section, we introduce building blocks for scE(match), namely, the scmap tool for computationally efficient single-cell cluster mapping and homomorphic encryption for privacy-preserving computations.
scmap: (Un-)supervised Projection of Single Cell Data
scmap [15] is a tool for projecting clusters from one query dataset into another reference dataset either on cell (cell-to-cell matching), or cluster-centroid level (cell-to-centroid matching). For integration, scmap employs a nearest neighbor search based on cosine similarity, Pearson correlation, and Spearman correlation and outputs a corresponding match if two of the three metrics exceed a certain threshold. In detail, the cell-to-centroid matching process that we will utilize later is designed as follows:
scmap identifies differentially-expressed genes (i.e., features) in the reference dataset either by assessing their variability [22] (unsupervised) or marker genes [16] (supervised) and then aligns the query dataset to the selected genes. Clusters in the reference dataset are then reduced to their centroid, i.e., the median of the cluster member’s features.
scmap computes the cosine similarity besides Pearson and Spearman correlation between each cell in the query dataset and cell/centroid in the reference dataset.
scmap considers a match if two of these three metrics consider a combination closest among the analyzed pairs and one metric exceeds an experimentally evaluated metric threshold. If neither combination of metrics exceeds the threshold, scmap treats a cell as unassigned.
Noteworthy, all these steps only involve computationally simple operations (sums, products, and exponentiation) [15]. While there is a quadratic complexity due to the pairwise combination of samples in the query and reference datasets, scmap converges in a defined number of steps that only depend on the input dimensionality. This behavior separates scmap from Harmony (which requires iterative optimization until convergence [17]) and deep-learning approaches like scVI (which require training and thus a potentially infinite number of steps [25]).
Homomorphic Encryption
Homomorphic Encryption (HE) is a cryptographic approach that enables computations on encrypted data without needing to decrypt the underlying raw data, thereby maintaining data confidentiality even during processing [40]. This capability makes HE particularly valuable for offloading computations to external (untrusted) third parties [41][33].
Over time, different HE cryptosystems emerged, each with distinct implications on expressiveness, performance, and usability [40]. These range from Partially Homomorphic Encryption (PHE) [42][43][44], which supports limited operations, over Somewhat Homomorphic Encryption (SWHE) [40], which allows for a predetermined number of operations with manageable overhead, to Fully Homomorphic Encryption (FHE) [45][46][47], which extends SWHE’s capabilities to unlimited operations but introduces a costly bootstrapping operation. Consequently, for scE(match), we envisage tailoring the HE cryptosystem to the required properties, specifically aiming at avoiding costly bootstrapping operations. Notably, the CKKS scheme [48] allows us to achieve FHE-level functionality for a fixed number of operations without the need for bootstrapping, thus approximating the performance of SWHE.
scE(match): Privacy-Preserving Cluster Matchings of Single-Cell Data
In this section, we introduce scE(match), our privacy-preserving adaptation of scmap for matching clustered single-cell data between two parties. We first provide an overview of the design, followed by a detailed description of the protocol flow between the two parties and the matching platform, the involved entities, and implementation-specific characteristics.
Design Overview
To enable a privacy-preserving cluster comparison between two clients $A$ and $B$ (see Section 3), we employ an adaption of scmap [15] that operates under HE to map a query dataset into a remote reference dataset. By doing so for both directions, we can aggregate the resulting mapping into a bilateral cluster-level matching that allows for identifying corresponding clusters between the two datasets. To this end, we replace the feature selection in scmap with a distributed, privacy-preserving variant and adapt the cell-to-centroid mapping step of scmap to operate on encrypted data.
Figure 3 provides an overview of the protocol flow between the two clients $A$ and $B$ and the matching platform. Specifically, we outsource the mapping to a third party, the mapping server, while keeping the sensitive genetic data confidential. We then employ another, from the mapping server separate party, the aggregation server, to exclusively hold decryption keys and thereby ensure data privacy. The aggregation server decrypts and aggregates calculated mappings from both parties and derives statistics, such as the number of commonly identified cell clusters. These statistics allow the clients $A$ and $B$ to match similarities between their datasets and findings without revealing the data or exclusively identified clusters to each other and shall be used to initiate further collaboration. The overall protocol flow is illustrated in Figure 3 and can roughly be divided into a preparation (Steps 1a to 5b), mapping (Steps 6a and 6b), and aggregation phase (Steps 7 to 10). In this protocol flow, the preparation and matching phases utilize our privacy-preserving adaptation of scmap, and the computation of matches can be considered a simple downstream analysis step on the integrated data. In the following, we provide a detailed description of each phase.
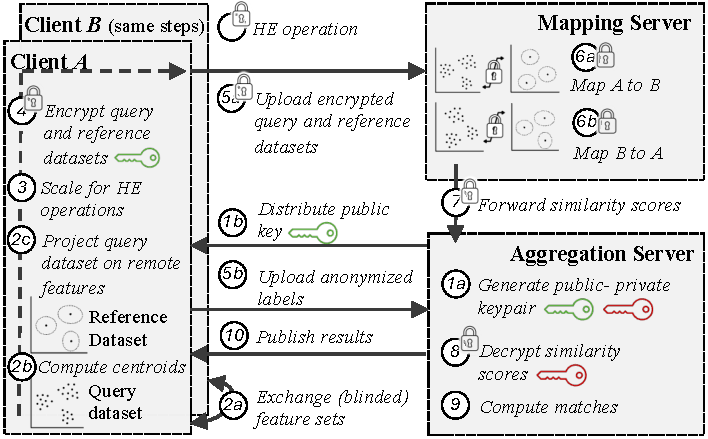
Preparation and Privacy-Preserving Feature Selection
Initially, the aggregation server generates a homomorphic encryption key pair (Step 1a) and distributes the public key to clients for data encryption (Step 1b). The clients then perform feature selection either via dropout [22] (unsupervised) or NS-Forest [16][49] (supervised), before communicating these selected features with the other client (Step 2a). Based on these features, the clients then compute centroids for their labels (Step 2b) and project their query dataset on the selected features of the remote client (Step 2c). In the unsupervised feature selection case on the one hand, we do not deem this selection sensitive, as it does not depend on the identified clusters, i.e., the data-provider’s IP. Clients $A$ and $B$ can thus exchange their selection without further protection. In the supervised feature selection case, on the other hand, the selected genes are marker genes for specific clusters that are supposed to differ between clusters. As these marker genes might hint at specific clusters being present in a remote dataset, we require the clients to exchange a blinded list of features by randomly sampling additional genes. We later show (cf. Section 6.4) that this blinding has negligible impact on the quality of calculated matchings.
Due to limitations in the SEAL homomorphic encryption library, which lacks support for square roots and division [50], and necessary comparison for calculating ranks, data must be preprocessed before encryption. For Pearson, we additionally need to subtract sample level means, and for Spearman, we calculate rank vectors before subtracting sample level means from the rank vector. Afterward, we must scale the precomputed vectors for all three metrics with their inverse Euclidean (L2) norm, such that a simple, homomorphically evaluated cross product yields cosine similarity, Pearson, and Spearman correlation. Consequently, clients must provide a pre-computed matrix (Step 3) for each metric rather than solely the cell$\times$gene/centroid matrix, balancing increased communication overhead with enhanced computational efficiency in homomorphic operations. The clients then encrypt (Step 4) and upload both datasets to the mapping server (Step 5a) before providing a permuted list of cluster indices per cell in the query dataset to the aggregation server (Step 5b), which is required to provide statistics on cell level later.
Privacy-Preserving Cluster Mapping
During this phase, the mapping server maps cells from the query dataset to the reference dataset based on selected features. scmap mappings, being directional from the query to the reference dataset, necessitate bi-directional matching. This matching involves projecting Client $A$’s data onto Client $B$’s dataset (Step 6a) and vice versa (Step 6b) by alternating the roles of the query and reference datasets. These computations are independent and can be executed concurrently or sequentially. To match the query to the reference, the mapping server calculates a dot product between each combination of samples from the query and reference dataset. The output from this step includes the calculated scores for all three metrics per cell and centroid in the query and reference datasets. The aggregation server then employs these matrices to analyze overlapping clusters between the two datasets.
Statistical Inference at the Aggregation Server
The aggregation server leverages the homomorphically computed mappings to generate statistics on overlapping clusters between the datasets of Clients $A$ and $B$. Therefore, it receives encrypted metric scores from the mapping server in Step 7, which it decrypts using its private key Step 8. This data comprises directional metric scores for each combination of cells and centroids in the query and reference datasets. Utilizing this information, the server evaluates each query dataset item in Step 9 to determine if it meets the matching criteria established by scmap [15], i.e., exceeding the threshold for at least two of the three metrics. The threshold’s strictness is adjustable based on client (mutually agreed) preferences. Subsequently, the server ranks clusters based on the count of matches from the opposing client’s dataset and shares these findings with both clients in Step 10. While the server primarily focuses on these match results to facilitate initial collaboration or further private data analysis, it can generate additional statistics, such as identifying split or highly overlapping clusters, if clients agree to share this information.
Entities and Operators
In scE(match), the primary entities are Clients $A$ and $B$, the mapping server, and the aggregation server. We assume Clients $A$ and $B$ will initiate the comparison out-of-band. As the number of laboratories working within the same research area is limited, they are likely familiar with each other. A third party operates the mapping server, which mitigates the need for mutual trust between clients regarding the accurate execution of the protocol. Conversely, the aggregation server is managed by a different third party. Non-collusion between the matching and aggregation servers is essential to prevent data leakage. Employing independent cloud providers for each server is a practical strategy to enforce this separation.
The protocol designates the mapping server as the most computationally demanding component, primarily handling data matching. In contrast, the aggregation server principally manages the download and decryption of matched results. Implementing a fee-for-service usage could motivate third-party operations. We suggest that research data management organizations, such as the German NFDI [51], would be ideal operators. These entities typically lack incentives to deviate from the protocol, and their reputation could suffer significantly if collusion is detected (cf. Section 3).
Implementation
We implemented scE(match) in Python, utilizing the Pyfhel library to interface with Microsoft SEAL’s CKKS homomorphic encryption (HE) scheme [50][48]. For handling single-cell data, we employed the h5ad format and scanpy, scipy, and numpy libraries for manipulation of pre-encryption data. Due to the original scmap algorithm being implemented in R, we re-implemented the necessary components in Python. A side-by-side comparison between the original scmap and our Python version yielded identical scores on one of the originally used datasets [52], confirming the accuracy of our implementation. As our focus is on computational efficiency, we simulated network communication by serializing data to disk. Moreover, we first implemented a single-threaded variant to assess its complexity regarding input data dimensions. To evaluate today’s state-of-the-art datasets, we also implemented a version that parallelizes data encryption and homomorphic computations (used for Section 6.4).
By design, CKKS supports a SIMD architecture, allowing multiple data points to be encrypted within a single ciphertext. We optimized the scmap algorithm to capitalize on this feature by encrypting data from up to eight cells or centroids into one ciphertext with 4096 slots. This optimization enables SIMD parallel processing of multiple cells or centroids, thereby reducing computational load by a factor of eight.
Evaluation
We now evaluate the precision and performance of scE(match) to show that
(i) the noise induced by homomorphic encryption does not impact the matching results, and (ii) the protocol is feasible for real-world applications, even on today’s large single-cell datasets.
| Dataset | Plaintext Data Size | # Cells | # Genes | # Types |
|---|---|---|---|---|
| D1 Litvinuková et al. [53] | 5.01 GB | 486,134 | 33,538 | 11 |
| D2 Chaffin et al. [54] | 13.5 GB | 592,689 | 36,601 | 21 |
Experimental Setup and Datasets
We conducted our evaluations on a single server (2x Intel Xeon Platinum 8160 CPU with 24 cores, hyper-threading, 192GB RAM, and an SSD). Each experiment was repeated 30 times to establish 95% confidence intervals. All processes were executed sequentially on the same server, with data transmission being simulated via disk storage.
Datasets Given the unavailability of original scmap datasets and the emergence of significantly larger single-cell datasets, we evaluated scE(match) using two recent datasets covering heart tissues, as shown in Table 1. As the control flow of HE protocols is independent of any data content, we do not expect significant runtime differences when utilizing other data. However, these datasets vary in cell and feature counts and exhibit varying clustering patterns, affecting the data volume and, thereby, overall runtime.
For the performance and storage evaluation, we subsample the Dataset D1 via stratified sampling to generate query datasets with an artificial number of cells. To also create datasets with an artificial number of clusters, we group clusters by their size and then perform stratified sampling from the top $n$ clusters. Here, we use five clusters with a total of 1000 cells as a baseline. As scmap recommends using 500 features, we fix this number by instructing our feature selection to select 500 genes.
Precision
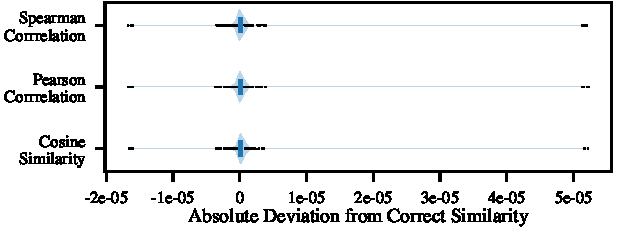
Given that CKKS ciphertexts approximate real numbers, we must assess whether this approximation substantially affects the matching results of scE(match). Here, any early imprecision might amplify in the following calculations. Figure 4 illustrates the difference in metric scores between plaintext scmap and scE(match). For all three metrics, 99% of scores calculated by scE(match) differ by less than 1.16×10^-06 from those of the pure scmap variant. Moreover, a worst-case analysis involving random perturbations of metric scores in $[0,1]$ indicates that 99% of metric score deviations that alter the matching decision, such as failing to recognize a genuine cluster member, deviate by at least 9.33×10^-03. Consequently, any imprecision introduced by HE is significantly lower than this threshold, ensuring that it does not compromise the matching accuracy of scE(match).
Performance and Storage
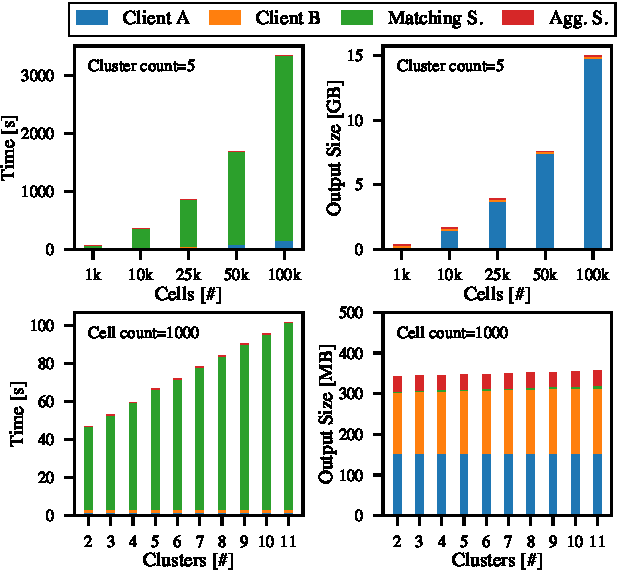
Initially, we conduct synthetic measurements of the complete protocol based on dataset characteristics such as the number of cells and clusters. As shown in Figure 5 (top), the analysis begins with a baseline dataset comprising 1000 cells and five clusters. We then increase the number of cells and clusters at one client to explore scE(match)’s scaling behavior (G4). Each step of the process and the computational demands for each entity are as follows.
Clients Clients start by reading and preprocessing datasets to optimize them for HE metric computations. This step includes data scaling and encryption before uploading the ciphertexts to the mapping server. For our largest synthetic dataset, i.e., 100000 cells, these steps consume 158 s $\pm$ 1.9, out of which the encryption is the most time-consuming operation with 142 s $\pm$ 1.6. The encryption results in three pre-scaled and encrypted versions of the feature matrix, requiring 14.8 GB of storage, an increase of 36.8 × compared to the plaintext dataset but still manageable with off-the-shelf hardware.
Mapping Server Upon receiving the encrypted data from clients, the mapping server calculates metric scores for each cell and centroid matrix combination in both matching directions. The runtime of this step is 3186 s $\pm$ 18 for the most extensive dataset. Again, our measurements indicate the homomorphic operations as the most resource-intensive consuming 3149 s $\pm$ 18, followed by data serialization and deserialization processes. The output of this step, i.e., the homomorphically encrypted metric scores require 51.2 MB of storage per dataset, an increase of 4.2 × compared to the plaintext dataset.
Aggregation Server The aggregation server’s role is to download the encrypted metric scores from the mapping server, decrypt them, and derive matching statistics for all cluster pairs between the two datasets. It, therefore, has received anonymized cluster indices from both clients and can attribute calculated cluster mappings to the correct clusters without revealing the cluster names. Out of the total runtime of 0.92 s $\pm$ 0.04 for these steps, the decryption of the metric scores is most time-intensive 0.16 s $\pm$ 0.01, whereas the rest is spent on data deserialization and serialization. However, the aggregation server’s requirements are minimal compared to the other entities, which is also reflected in the output size of 35.9 MB.
Scalability The dependency of the runtime on each number of cells and clusters is linear, as shown in Figure 5. Specifically, doubling the amount of cells also increases runtime by a factor of two. This ratio is even better for the number of clusters, as we observe a doubling in runtime when increasing the cluster count by nine. As today’s single-cell datasets mostly scale in the number of cells but rarely exceed 20 different meaningful clusters [20], we derive a mostly linear scaling regarding dataset sizes.
Today’s single-cell datasets are typically well beyond the cell counts evaluated in Figure 5. To evaluate larger datasets, we thus employ a parallelized version of our implementation (cf. Section 5.6), which distributes de/encryption and homomorphic computation across the up to 96 threads of our server. For the 100000 cell dataset measured in Figure 5, this parallelized version reduces the runtime by a factor of 12.7 compared to the sequential version. This speedup is sourced mainly by the parallelizability of the client-side encryption (three precomputed matrices can be encrypted in parallel) and server-side mapping operations (ciphertext batches can be processed in parallel at the cost of duplicating the reference dataset). Additional startup costs for small datasets dominate the speedup but tend to reduce for large datasets. For the full version of Datasets D1 and D2, Table 2 provides an overview of the runtime and storage requirements, utilizing the paralellized implementation. Overall, we find that scE(match)’s performance renders it well-suited for real-world applications, even on today’s large-scale single-cell datasets. In the following, we show that scE(match) also provides useful matching results on these datasets, which promise to support research in the field.
| Dataset | FS | Runtime | Output [GB] (CA/CB/M/A) | Prec. [%] | Rec. [%] | Unassig. [%] |
|---|---|---|---|---|---|---|
| D1 | U | 34m 45s | 33/33/0.5/0.001 | 74/73 | 51/52 | 25/25 |
| S | 7m 33s | 8.3/8.3/0.5/0.001 | 81/81 | 82/81 | 5.6/6.6 | |
| SB | 16m 8s | 17/17/0.5/0.001 | 81/81 | 81/78 | 7.1/10 | |
| D2 | U | 1h 21m 30s | 44/44/1.2/0.001 | 90/90 | 83/83 | 6.3/6.7 |
| S | 37m 38s | 22/22/1.2/0.001 | 91/92 | 91/92 | 1.7/1.6 | |
| SB | 1h 21m 21s | 44/44/1.2/0.001 | 90/91 | 90/91 | 2.9/2.5 | |
| D1 ↔ D2 | U | 1h 46m 41s | 67/87/1.5/0.001 | 87/85 | 57/48 | 32/43 |
| S | 38m 8s | 33/22/1.5/0.001 | 86/86 | 76/77 | 13/12 | |
| SB | 1h 22m 17s | 67/44/1.5/0.001 | 86/87 | 70/71 | 20/20 |
Real-World Utility
To demonstrate that scE(match) facilitates privacy-preserving clustering comparisons of real-world datasets, we evaluate its utility (G3) on two distinct datasets. These datasets were independently collected and analyzed by different research groups, featuring inherent batch effects and hardware biases, thus mirroring the intended application scenario for scE(match).
Datasets After using the Dataset D1 as a basis for subsampling in our synthetic performance evaluation, we now study the entire Dataset D1, as well as a second Dataset D2 which also covers heart tissues. While Dataset D1 features different cell types from healthy donors, Dataset D2 also contains samples from patients with hypertrophic (HCM) and dilated cardiomyopathy (DCM) [54], which do not have corresponding clusters in the D1 dataset. Besides, both datasets contain partly overlapping cell types that can considered similar. For instance, D1 includes “Pericytes”, whereas D2 includes “Pericytes_I” and “Pericytes_II”. To assess matching accuracy, a domain expert created a baseline mapping of these cell types, which serves as ground truth for our evaluation of the cross-projection scenario. Here, an ideal matching between D1 and D2 (with precision and recall equal to one) would project corresponding cell types on each other and map remaining cell types to “unassigned” in the other dataset.
Matching Results Table 2 presents the matching outcomes for entire datasets in both self-projection and cross-projection scenarios. We observe that scE(match) accurately identifies most relationships. Supervised feature selection consistently achieves the highest precision and recall, while its unsupervised counterpart tends to leave a higher proportion of cells unassigned. In the supervised-blinded variant, designed to prevent direct transmission of marker genes to the other client by adding a randomly selected feature for each marker gene, there is an increase in unassigned cells and a reduction in recall. However, scores remain closer to supervised than to unsupervised feature selection. Besides, precision remains robust. This supports the effectiveness of blinding as a strategy to obscure specific marker genes.
Supervised Feature Selection Reduces Runtime A side effect of supervised feature selection can be a reduction of the selected features for the metric computation. Compared to the 500 features of unsupervised feature selection, in our case, supervised feature selection yields between 92 and 173 features which depends on the dataset, dataset size, and random initialization of NS-Forest. As shown in Table 2, this decrease leads to a reduction in runtime by 78% for D1, 54% for D2 and 64% for D1$\leftrightarrow$D2, compared to unsupervised feature selection and with similar effects on client output sizes.
Security and Privacy Discussion
We assume the HE cryptosystem to be secure; as such, it will ensure data privacy. Nonetheless, our protocol execution might expose certain information about data and metadata. We detail these aspects and discuss their implications for the privacy (G1) and IP (G2) of data providers, and propose potential mitigation strategies. Additionally, we revisit our security assumptions, particularly concerning honest-but-curious entities and the non-collusion requirement between the matching and aggregation servers (cf. Section 3), and discuss the consequences of relaxing these requirements.
Metadata Leakage The encrypted dataset size reveals the approximate number of cells and centroids to the matching and aggregation servers (cf. Figure 5). As clients anticipate some information disclosure as part of the protocol, we consider this leakage acceptable. Moreover, despite being data-independent, our protocol does not conceal any communication patterns, and it reveals the identities of entities and their communication frequency. Although not considered sensitive, these patterns could be obscured using mechanisms like Tor [55].
Mapping Granularity While cluster names need not be visible to remote clients (they only see anonymized indices), this complete mapping might still have privacy implications: For instance, information as shown in Figure 2 might reveal when clusters have been further subclustered in a remote dataset, and we suppose that repeatedly adding artificially placed cells in the query dataset or additional centroids in the reference dataset will eventually help to triangulate specific cell clusters. As a mitigation strategy, the clients could agree to apply thresholding to the mapping results. Then, the aggregation server would, for instance, only reveal a relation to the largest matching cell cluster from the query dataset. For the exemplary mapping shown in Figure 2, such a result would, for instance, exclusively map “Pericyte I” to the “Pericytes” class of the other dataset, while disregarding all other mappings that originate from the “Pericyte I” cluster. We thus consider filtering a viable countermeasure to reduce leaking such sensitive IP.
Non-Collusion Removing the non-collusion requirement risks severe privacy breaches, such as decrypting precomputed matrices to infer raw gene expression data. Similarly, collusion between a client and the aggregation server could expose detailed metric scores, potentially enabling triangulation of data positions in the other client’s dataset, thus infringing on intellectual property rights. However, a client must collude with the matching and aggregation servers to access the other client’s raw data. We thus consider this threat to be impractical in our setting (cf. Section 3).
Iterative Protocol Execution Our protocol is designed to only provide matching results without detailed correlation scores at the cellular level. We suppose that frequent re-evaluation of scE(match) while slightly shifting cells in the query datasets eventually allows some triangulation of the centroids in the reference dataset. A single execution of the protocol, however, can only leak this information on a coarse level, i.e., for the locally provided centroids. As protocol execution requires consent from both clients, we do not consider this attack vector practically relevant.
Overall, we assess the security and privacy implications of scE(match) to be manageable for the intended applications.
Discussion and Impact
scE(match) provides single-cell researchers with a tool for privacy-preserving comparison and integration of sensitive datasets while maintaining data confidentiality. It demonstrates scalability beyond today’s single-cell datasets, delivers precise matching results, and facilitates collaboration on previously inaccessible data. We now explore this research’s implications, limitations, and future directions.
Impact scE(match) offers insights into how various researchers categorize their cell datasets, comparing annotations, cell types, and other cluster-dependent information. Typically, sharing such detailed information requires a lengthy publication process or might not occur if findings are inconclusive. Here, scE(match) constitutes a novel tool that accelerates the initiation of new collaborations across laboratories, especially where privacy and intellectual property previously impeded such efforts. It thus constitutes an ideal tool for privacy-concerned researchers.
Limitations and Alternative Designs scE(match) may face adoption challenges due to its runtime overhead factor of 46.2 compared to plaintext execution and the need for two third parties for matching and aggregation. Subsampling the query dataset might be one option to decrease runtime. Alternatively, moving the matching process to clients poses minimal security risks, allowing data to be archived locally while waiting for potential future compromises of the cryptosystem or keys. If clients are willing to share their encrypted centroids with each other, the mapping server can thus be omitted. This approach eliminates the need for a mapping server by directly sharing encrypted data among clients, significantly reducing the observed communication overhead by scE(match).
Future Work As our main objective was to show the feasibility of scE(match), this study did not evaluate the networking components. To ensure its practical applicability as a ready-to-use tool, these components should be implemented and tested in real-world settings despite anticipated minimal communication overhead. Besides, future improvements could involve adapting existing tools like Harmony, which have surpassed scmap in cell-level integration tasks [17][16], to support homomorphic operations for secure data integration and downstream analysis not only on the cluster but also on cell level. Here, cell-level integration would enable the privacy-preserving adaptation of more advanced downstream analysis tasks, such as joint derivation of annotations with increased statistical power. Finally, future work should aim at extended the biological utility of scE(match) by considering more, and more diverse datasets, such as extending the evaluation to datasets from and across different tissues or organisms.
Overall, we are the first to add privacy preservation to cluster-to-cluster matching in single-cell research. Thus, we provide significant added value for collaborating on private single-cell datasets.
Conclusion
In this study, we introduced scE(match), a privacy-preserving tool designed to address significant challenges in collaborative single-cell RNA sequencing data analysis. By leveraging homomorphic encryption, scE(match) enables the secure comparison of single-cell clusters between different datasets. Our comprehensive evaluation demonstrates that scE(match) can accurately match clustered cells across disparate datasets while maintaining the confidentiality and integrity of the data. This mitigates risks related to data privacy and IP leakage and facilitates a collaborative environment where researchers can securely compare findings and accelerate scientific discoveries. Although preliminary, our promising results highlight its potential, with future enhancements planned for performance optimization and real-world testing of its networking components.
Ethics Declarations
Since the evaluation of scE(match) relies on published datasets [53][54], our research did not directly involve human subjects or their samples. Consequently, for our work, we did not require IRB approval or consent from the participants.
References
Spatially resolved multiomics of human cardiac niches,Nature, vol. 619, no. 7971, 2023.
An atlas of healthy and injured cell states and niches in the human kidney,Nature, vol. 619, no. 7970, 2023.
Decoding myofibroblast origins in human kidney fibrosis,Nature, vol. 589, no. 7841, 2021.
A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications,Genome Med., vol. 9, no. 1, 2017.
The technological landscape and applications of single-cell multi-omics,Nat. Rev. Mol. Cell Biol., vol. 24, no. 10, 2023.
Dissecting CD8+ T cell pathology of severe SARS-CoV-2 infection by single-cell immunoprofiling,Front. Immunol., vol. 13, 2022.
Refining colorectal cancer classification and clinical stratification through a single-cell atlas,Genome Biol., vol. 23, no. 1, 2022.
Spatial multi-omic map of human myocardial infarction,Nature, vol. 608, no. 7924, 2022.
Single-cell multi-omic and spatial profiling of human kidneys implicates the fibrotic microenvironment in kidney disease progression,Nat. Genet., vol. 56, no. 8, 2024.
Resolving Individuals Contributing Trace Amounts of DNA to Highly Complex Mixtures Using High-Density SNP Genotyping Microarrays,PLoS Genet., vol. 4, no. 8, 2008.
Overcoming barriers to single-cell RNA sequencing adoption in low- and middle-income countries,Eur. J. Hum. Genet., vol. 32, no. 10, 2024.
Exponential scaling of single-cell RNA-seq in the past decade,Nat. Protoc., vol. 13, no. 4, 2018.
The FAIR Guiding Principles for scientific data management and stewardship,Sci. Data, vol. 3, 2016.
Eleven grand challenges in single-cell data science,Genome Biol., vol. 21, no. 1, 2020.
scmap: projection of single-cell RNA-seq data across data sets,Nat. Methods, vol. 15, no. 5, 2018.
FR-Match: robust matching of cell type clusters from single cell RNA sequencing data using the Friedman–Rafsky non-parametric test,Brief. Bioinform., vol. 22, no. 4, 2021.
Fast, sensitive and accurate integration of single-cell data with Harmony,Nat. Methods, vol. 16, no. 12, 2019.
scGPT: toward building a foundation model for single-cell multi-omics using generative AI,Nat. Methods, vol. 21, no. 8, 2024.
Deep Learning in Single-cell Analysis,ACM Trans. Intell. Syst. Technol., vol. 15, no. 3, 2024.
M3Drop: dropout-based feature selection for scRNASeq,Bioinformatics, vol. 35, no. 16, 2019.
Comprehensive Integration of Single-Cell Data,Cell, vol. 177, no. 7, 2019.
Probabilistic harmonization and annotation of single‐cell transcriptomics data with deep generative models,Mol. Syst. Biol., vol. 17, no. 1, 2021.
Deep generative modeling for single-cell transcriptomics,Nat. Methods, vol. 15, no. 12, 2018.
scGen predicts single-cell perturbation responses,Nat. Methods, vol. 16, no. 8, 2019.
Inferring Genetic Ancestry: Opportunities, Challenges, and Implications,Am. J. Hum. Genet., vol. 86, no. 5, 2010.
Correlation between Genetic and Geographic Structure in Europe,Curr. Biol., vol. 18, no. 16, 2008.
Identifying Personal Genomes by Surname Inference,Science, vol. 339, no. 6117, 2013.
scFed: federated learning for cell type classification with scRNA-seq,Brief. Bioinf., vol. 25, no. 1, 2023.
Privacy-preserving integration of multiple institutional data for single-cell type identification with scPrivacy,Sci. China Life Sci., vol. 66, no. 5, 2023.
PPML-Omics: A privacy-preserving federated machine learning method protects patients’ privacy in omic data,Sci. Adv., vol. 10, no. 5, 2024.
BLOOM: BLoom filter based Oblivious Outsourced Matchings,BMC Medical Genom., vol. 10 (Suppl 2), 2017.
Secure genome-wide association analysis using multiparty computation,Nat. Biotechnol., vol. 36, no. 6, 2018.
Practical Privacy-Preserving K-means Clustering,PoPETS, vol. 2020, no. 4, 2020.
Privacy-preserving Density-based Clustering,In Proc. ACM ASIACCS, 2021
Scalable Differentially Private Clustering via Hierarchically Separated Trees,In Proc. ACM KDD, 2022
They who must not be identified—distinguishing personal from non-personal data under the GDPR,Int. Data Priv. Law, vol. 10, no. 1, 2020.
A Survey on Homomorphic Encryption Schemes: Theory and Implementation,ACM Comput. Surv., vol. 51, no. 4, 2018.
CryptDB: Protecting Confidentiality with Encrypted Query Processing,In Proc. ACM SOSP, 2011
A Method for Obtaining Digital Signatures and Public-key Cryptosystems,Commun. ACM, vol. 21, no. 2, 1978.
Probabilistic encryption,J. Comput. Syst. Sci., vol. 28, no. 2, 1984.
Public-Key Cryptosystems Based on Composite Degree Residuosity Classes,In Proc. EUROCRYPT, 1999
Fully Homomorphic Encryption Using Ideal Lattices,In Proc. ACM STOC, 2009
Fully Homomorphic Encryption over the Integers,In Proc. EUROCRYPT, 2010
Survey on Fully Homomorphic Encryption, Theory, and Applications,Proc. IEEE, vol. 110, no. 10, 2022.
Homomorphic Encryption for Arithmetic of Approximate Numbers,In Proc. ASIACRYPT, 2017
Nationale Forschungsdateninfrastruktur (NFDI),Inform. Spektrum, vol. 44, no. 5, 2021.
Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells,Nat. Struct. Mol. Biol., vol. 20, no. 9, 2013.
Cells of the adult human heart,Nature, vol. 588, 2020.
Single-nucleus profiling of human dilated and hypertrophic cardiomyopathy,Nature, vol. 608, no. 7921, 2022.
On Anonymity in an Electronic Society: A Survey of Anonymous Communication Systems,ACM Comput. Surv., vol. 42, no. 1, 2009.
@inproceedings{johannes-lohmoeller2024sce(match):,
title = {scE(match): Privacy-Preserving Cluster Matching of Single-Cell Data},
author = {johannes-lohmoeller and Jannis Scheiber and Rafael Kramann and Klaus Wehrle and Sikander Hayat and Jan Pennekamp},
booktitle = {Proceedings of the 23rd IEEE International Conference on Trust, Security and Privacy in Computing and Communications},
year = {2024},
doi = {10.1109/TrustCom63139.2024.00294},
}